Research Published at UMBC
Want to know more? Follow us on Twitter: @BennettLabsUMBC
65. Adsorption-Induced Ferroelectric Symmetry Breaking in Two-Dimensional CuInP2S6
Peng Yan and Joseph W. Bennett, ACS Appl. Mater. Inter., 2026 (link)
64. Insulating A2VX3 Oxides and Sulfides as Ferromagnetic Wires
Anthony Casale and Joseph W. Bennett, Phys. Rev. Mater., 2026, 10, 064412-1-11 (link)
63. Chiral molecular intercalation enables light-controlled 2D multiferroic heterostructures
Wang, Zhongxuan; Hu, Yong; Fang, Zhenyao; Wang, Yixiao; Yan, Peng; Sun, Jiayue; Lopez, Mario; Stadel, Ryan; Niu, Yuchen; Little, Joshua; Yan, Qimin; Chen, Po-Yen; Gong, Cheng; Bennett, Joseph; Gu, Genda; Li, Qiang; Chen, Zhijie; Woehl, Taylor; Rodriguez, Efrain; Liu, Kai; and Ren, Shenqiang, Nano Letters, 2026 (in press, link)
62. Modeling 2D van der Waals Materials with Homonuclear Bonds of Main Group Cations
Peng Yan, Anthony Casale, and Joseph W. Bennett, ACS Organic and Inorganic Au, 2026, 6, 104-118 (link)
61. Compositional Design Rules for Tuning Functionalities in CuInP₂X₆ (X=S, Se) Van der Waals Semiconductor Ferroelectrics
Mona Layegh and Joseph W. Bennett, Dalton Trans., 2025, 54, 14384-14395 (link)
60. Data-Enabled Discovery of Two-Dimensional van der Waals Layered Phosphochalcogenides
Peng Yan, Mona Layegh, Ryan Stadel, Joshua Birenzvige, Peter Y. Zavalij, Efrain E. Rodriguez, and Joseph W. Bennett
Chem. Mater., 2025, 37, 14, 5086–5098 (link)

59. Evidence of ferrimagnetism in Fe3GaTe2 via neutron diffraction studies
Mario Lopez, Peng Yan, Peter Y. Zavalij, Anahita Javadi, Ivan da Silva, Zhongxuan Wang, Shenqiang Ren, Joseph W. Bennett, and Efrain E. Rodriguez
J. Mater. Chem. C., 2025 (link)
58. Oleic acid rearrangement enables facile transfer of red-emitting quantum dots from hexane into water with enhanced fluorescence
Tohid Baradaran, Jasper Tucker, Chanda M. Lowrance, Lekan Ajiboye, Matthew Pelton, Joseph W. Bennett, and Marie-Christine Daniel
Nanoscale, 2025, 17, 12894-12910 (Link)
57. Open-Source DFT Calculations of Electronic Structure to Understand Bonding in Solids
Mona Layegh and Joseph W. Bennett
J. Chem. Ed., 2025, 102, 5, 1803–1813 (Link)
56. Assessing the K2BO3 Family of Materials as Multiferroics
Anthony A. Casale and Joseph W. Bennett
Physical Review Materials, 2024 (8) 114424
https://journals.aps.org/prmaterials/pdf/10.1103/PhysRevMaterials.8.114424

55. The Formation and Stability of 3D and 2D Materials
Mona Layegh, Peng Yan, Joseph W. Bennett
Progress in Crystal Growth and Characterization of Materials, 2024 (70), 100615
https://doi.org/10.1016/j.pcrysgrow.2023.100615
54. The Effects of Chlorine-Containing Species on Cinnabar: A Density Functional Theory Investigation into the Surface Adsorption Reactivity of Mercury Sulfide
Aria Tauraso, G. Amalthea Trobare, Lillian G. Kidd, Jessica E. Heimann, Zeev Rosenzweig, Joseph W. Bennett*
Surface Science, 2024 (740), 122412
https://doi.org/10.1016/j.susc.2023.122412
*Featured in the Young Investigator Special Issue 2023
53. Chemical Transformations of 2D Kaolinic Clay Mineral Surfaces from Sulfuric Acid Exposure
Chari, Celia; Heimann, Jessica; Rosenzweig, Zeev; Bennett, Joseph W.*; Faber, Katherine
Langmuir, 2023 (39), 6964-6974
https://pubs.acs.org/doi/full/10.1021/acs.langmuir.3c00113
52. Understanding the Effects of Amine and Morpholine Adsorption on Unglazed Earthenware using Density Functional Theory
Jessica E. Heimann, Zeev Rosenzweig, Joseph W. Bennett*
J. Cult. Herit., 2023 (61) 168-176
https://doi.org/10.1016/j.culher.2023.04.002
51. A DFT Combined with Thermodynamics Exploration of Novel 2D Materials Created Using Aqueous Exfoliation
Mona Layegh and Joseph W. Bennett*
J. Phys. Chem. C., 2023 (127) 2314-2325
https://pubs.acs.org/doi/full/10.1021/acs.jpcc.2c08053
*Special issue “Early Career and Emerging Researchers in Physical Chemistry”
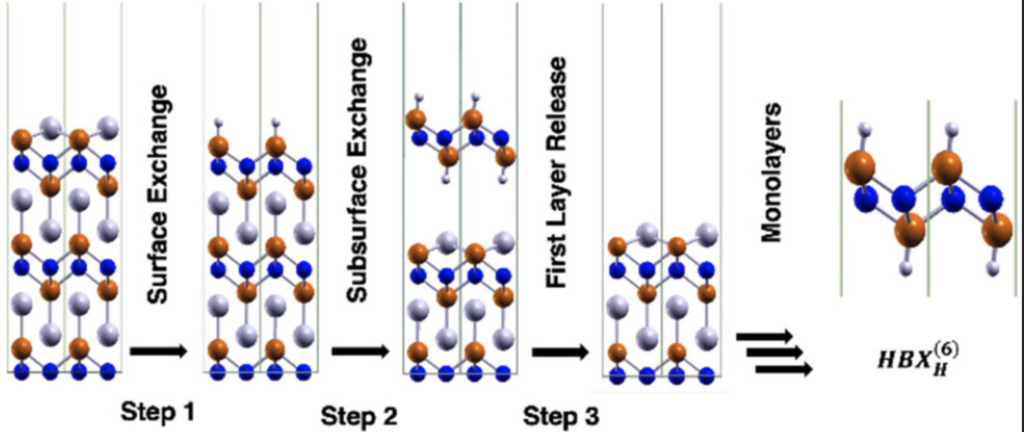
50. Surface Transformation Thermodynamics of Alkaline Earth Carbonates using First-Principles Calculations
Ryan T. Grimes and Joseph W. Bennett*
Surface Science, 2022 (726) 122165 https://doi.org/10.1016/j.susc.2022.122165
*Selected for the cover of the December 2022 issue of Surface Science
49. Giant and Controllable Photoplasticity and Photoelasticity in Compound Semiconductors
Jiahao Dong, Yifei Li, Yuying Zhou, Alan Schwartzman, Haowei Xu, Bilal Azhar, Joseph Bennett, Ju Li, and R. Jaramillo
Phys. Rev. Lett., 2022, (129), 065501
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.129.065501
*Highlighted in Physics as an Editor’s Spotlight: https://physics.aps.org/articles/v15/s102
48. A Density Functional Theory (DFT) Investigation of Sulfur-Based Adsorbate Interactions on Alumina and Calcite Surfaces
Stanley Ou, Jessica E. Heimann; Joseph W. Bennett*
Clays and Clay Minerals (2022)
https://link.springer.com/article/10.1007/s42860-022-00194-5
47. Density Functional Theory (DFT) as a Non-Destructive Probe in the Field of Art Conservation: Small Molecule Adsorption on Aragonite Surfaces
Heimann, Jessica; Tucker, Jasper; Huff, Layla; Kim, Ye Rin; Ali, Jood; Stroot, M. Kaylor; Welch, Xavier; White, Harley; Wilson, Marcus; Wood, Cecelia; Gates, Glenn; Rosenzweig, Zeev; Bennett, Joseph*
ACS Appl. Mater. Inter., 2022, (14), 13858-13871
https://pubs.acs.org/doi/abs/10.1021/acsami.1c23695
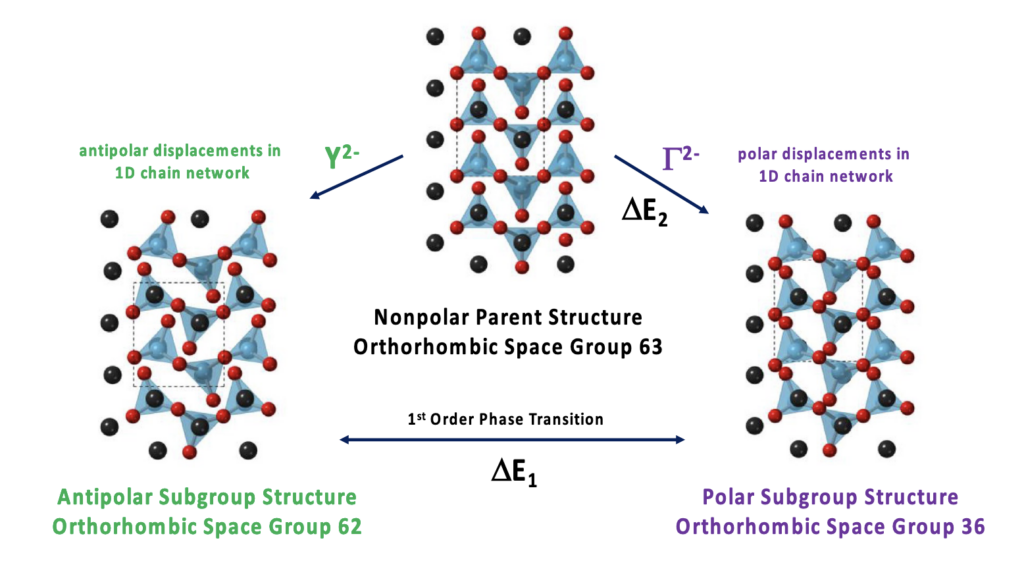
46. Developing New Antiferroelectric and Ferroelectric Oxides and Chalcogenides Within the A2BX3 Family*
A. C. Khan, A. S. Cook, J. A. Leginze, and J. W. Bennett*
J. Mater. Res., 2022, (37), 346-359
https://link.springer.com/article/10.1557/s43578-021-00410-3
*Special Issue Highlighting Early Career Materials Scientists 2022
45. Baltimore SCIART: A Fully Virtual Undergraduate Research Experience at the Interface of Computational Chemistry and Art
J. E. Heimann, T. H. Williams, J. W. Bennett, and Z. Rosenzweig
J. Chem. Ed., 2021, (98) 3172-3179
https://pubs.acs.org/doi/10.1021/acs.jchemed.1c00425
44. A Density Functional Theory (DFT) Investigation of How Small Molecules and Atmospheric Pollutants Relevant to Art Conservation Adsorb on Kaolinite
J. E. Heimann, R. T. Grimes, Z. Rosenzweig, and J. W. Bennett*
Appl. Clay Sci., 2021 (206) 106075
https://www.sciencedirect.com/science/article/pii/S0169131721000995?dgcid=author
43. Surface Transformations of Lead Oxides and Carbonates Using First-Principles and Thermodynamics Calculations
R. T. Grimes, J. A. Leginze, R. Zochowski, and J. W. Bennett*
Inorg. Chem., 2021 (60) 1228-1240
https://pubs.acs.org/doi/10.1021/acs.inorgchem.0c03398
* Featured in the “Out in Inorganic Chemistry: A Celebration of LGBTQIAPN+ Inorganic Chemists” Issue of Inorganic Chemistry
Out in Inorganic Chemistry Special Issue
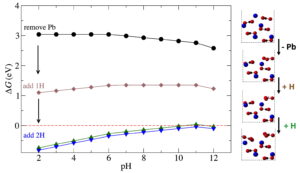
42. Exploring the A2BX3 Family for New Functional Materials using Crystallographic Database Mining and First-Principles Calculations
J. W. Bennett*, J. Phys. Chem. C., 2020
https://pubs.acs.org/doi/10.1021/acs.jpcc.0c03093
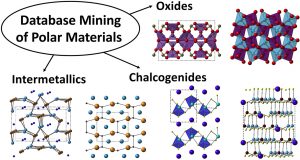
41. Surveying Polar Materials in the Inorganic Crystal Structure Database
to Identify Emerging Polar Structure Types
J. W. Bennett*, J. Solid State Chem., 2020 (281) 121045
https://www.sciencedirect.com/science/article/pii/S002245961930550X
Research Published at the University of Iowa
40. Understanding the Mechanism of Secondary Cation Release from the (001) Surface of Li(Ni1/3Mn1/3Co1/3)O2: Insights from First-Principles; Blake G. Hudson, Diamond T. Jones,Victoria M. Rivera Bustillo, Joseph W. Bennett and Sara E. Mason
J. Phys. Chem. C., 127, 43, 21022–21032 (2023)
https://pubs.acs.org/doi/10.1021/acs.jpcc.3c02764
39. Examining the Aufbau Principle and Ionization Energies: A Computational Chemistry Exercise for the Introductory Level
I. K. Metz, J. W. Bennett, and S. E. Mason
J. Chem. Ed., 98, 4017-4025 (2021)
https://pubs.acs.org/doi/abs/10.1021/acs.jchemed.1c00700
38. Density functional theory and thermodynamics analysis of MAl12 Keggin substitution reactions: Insights into ion incorporation and experimental confirmation
J. L. Bjorklund, M. Shohel, J. W. Bennett, M. E. Carolan, J. A. Smith, E. Holler, T. Z. Forbes and S. E. Mason
J. Chem. Phys., 154, 064303 (2021)
https://aip.scitation.org/doi/10.1063/5.0038962
37. First-principles and Thermodynamics Comparison of Compositionally-Tuned Delafossites: Cation Release from the (001) Surface of Complex Metal Oxides
J. W. Bennett, D. T. Jones, B. G. Hudson, J. Melendez-Rivera, R. J. Hamers and S. E. Mason
Environ. Sci.: Nano, 2020 (7) 1642-1651
https://doi.org/10.1039/C9EN01304K
36. DFT and Thermodynamics Calculations of Surface Cation Release in LiCoO2
A. Abbaspour-Tamijani, J. W. Bennett, D. T. Jones, N. Cartagena-Gonzalez, Z. R. Jones, E. D. Laudadio, R. J. Hamers, J. A. Santana and S. E. Mason
Applied Surface Science, 2020 (515) 145865
https://doi.org/10.1016/j.apsusc.2020.145865
35. Nickel enrichment of next-generation NMC nanomaterials alters material stability, causing unexpected dissolution behavior and observed toxicity to S. oneidensis MR-1 and D. magna
J. T. Buchman, E. A. Bennett, C. Wang, A. Abbaspour-Tamijani, J. W. Bennett, B. G. Hudson, C. M. Green, P. L. Clement, B. Zhi, A. H. Henke, E. D. Laudadio, S. E. Mason, R. J. Hamers, R. D. Klaper and C. L. Haynes
Environ. Sci.: Nano, 2020 (7) 571-587
https://pubs.rsc.org/en/content/articlelanding/2020/en/c9en01074b
34. A Systematic Determination of Hubbard U using the GBRV Ultrasoft Pseudopotential Set
J. W. Bennett, B. G. Hudson, I. Metz, D. Liang, S. Spurgeon, Q. Cui and S.E. Mason
Computational Materials Science, 2019 (170) 109137
https://www.sciencedirect.com/science/article/pii/S0927025619304288
33. Modeling of MAl12 Keggin Heteroatom Reactivity by Anion Adsorption
J. L. Bjorklund, J. W. Bennett, T. Z. Forbes and S. E. Mason
Crystal Growth & Design, 2019 (19) 2820-2829
https://pubs.acs.org/doi/abs/10.1021/acs.cgd.9b00044

32. DFT Computed Dielectric Response and THz Spectra of Organic Co-Crystals and Their Constituent Components
J. W. Bennett, M. E. Raglione, S. M. Oburn, L. M. MacGillivray, M. A. Arnold and S. E. Mason
Molecules, 2019 (24) 959
https://www.mdpi.com/1420-3049/24/5/959
31. Methane Dissociation on alpha-Fe2O3(0001) and Fe3O4(111) Surfaces:
First-Principles Insights into Chemical Looping Combustion
J. W. Bennett, X. Huang, Y. Fang, D. M. Cwiertny, V. H. Grassian and S. E. Mason
J. Phys. Chem. C., 2019 (123) 6450-6463
https://pubs.acs.org/doi/abs/10.1021/acs.jpcc.8b08675
30. Dissolution of Compositionally-Tuned Complex Metal Oxides: A First-Principles and Thermodynamics
Study of Cation Removal From the (001) Surface of Mn-rich Lithium Nickel Manganese Cobalt Oxide
J. W. Bennett, D. Jones, R. J. Hamers, and S. E. Mason
Inorg. Chem., 2018 (57) 13300-13311
https://pubs.acs.org/doi/abs/10.1021/acs.inorgchem.8b01855
29. Impact of Phosphate Adsorption on Complex Lithium Cobalt Oxide Nanoparticle Dispersibility in Aqueous Media
E. D. Laudadio, J. W. Bennett, C. M. Greene, S. E. Mason and R. J. Hamers
Environ. Sci. Technol., 2018 (52) 10186-10195
https://pubs.acs.org/doi/abs/10.1021/acs.est.8b02324
28. The Dissolution of Complex Metal Oxides from First-Principles and Thermodynamics: Cation Removal from the (001) Surface of Li(Ni1/3Mn1/3Co1/3)O2
J. W. Bennett, D. Jones, X. Huang, R. J. Hamers and S. E. Mason
Environ. Sci. Technol., 2018 (52) 5792-5802
https://pubs.acs.org/doi/abs/10.1021/acs.est.8b00054

27. Analysis of Conformational Properties of Amine Ligands at the Gold/Water Interface with QM, MM,
and QM/MM simulations *Selected as a PCCP HOT Article
D. Liang, J. Hong, D. Fang, J. W. Bennett, S. E. Mason, R. J. Hamers and Q. Cui
Phys. Chem. Chem. Phys., 2018 (20) 3349-3362
https://pubs.rsc.org/en/content/articlehtml/2018/cp/c7cp06709g
26. A Survey of the Reactivity Relationships of Anionic Adsorbates on Aluminum Nanoclusters
J. W. Bennett, J. L. Bjorklund, T. Z. Forbes and S. E. Mason
Inorg. Chem., 2017 (56) 13014-13028
https://pubs.acs.org/doi/abs/10.1021/acs.inorgchem.7b01803

25. Research highlights: comparing the biological response of nanoparticle solid solutions
J. W. Bennett, C. Allen, S. Pramanik, M. J. Gallagher, N. V. Hudson-Smith, D. Jones, M. O. P. Krause and S. E. Mason
Environ. Sci.: Nano, 2017 (4) 1428-1432
https://pubs.rsc.org/en/content/articlehtml/2015/en/c7en90025b
24. Ab initio Atomistic Thermodynamics Study of the (001) Surface of LiCoO2in a Water Environment and
Implications for Reactivity under Ambient Conditions
X. Huang, J.W. Bennett, M. N. Hang, E. D. Laudadio, R. J. Hamers, and S. E. Mason
J. Phys. Chem. C., 2017 (121) 5069-5080
https://pubs.acs.org/doi/abs/10.1021/acs.jpcc.6b12163
23. Influence of Nickel Manganese Cobalt Nanoparticle Composition on Toxicity Toward Shewanella
Oneidensis MR-1: Redesigning for Reduced Biological Impact
I. L. Gunsolus, M. N. Hang, N. V. Hudson-Smith, J. Buchman, J. W. Bennett, D. Conroy, S. E. Mason, C. Haynes and R. Hamers
Environ. Sci.: Nano, 2017 (4) 636-646
22. Systematic Density Functional Theory Study of the Structural and Electronic Properties of Constrained
and Fully Relaxed (001) Surfaces of Alumina and Hematite
K. W. Corum, X. Huang, J. W. Bennett and S. E. Mason
Molec. Simul. 2017 (43) 406-419 (Special Issue on Surface Chemistry)
https://www.tandfonline.com/doi/abs/10.1080/08927022.2017.1285402
Postdoctoral Research Published at Rutgers University
21. First-Principles Bulk-Layer Model for Dielectric and Piezoelectric Responses in Superlattices
J. Bonini, J. W. Bennett, P. Chandra and K. M. Rabe
Phys. Rev. B., 2019 (99) 104107
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.99.104107
20. Antiferroelectric topological insulators in ABC compounds
B. Monserrat, J. W. Bennett, K. M. Rabe, and D. Vanderbilt
Phys. Rev. Lett., 2017 (119) 036802
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.119.036802
19. Pseudopotentials for high-throughput DFT calculations:
K. F. Garrity, J. W. Bennett, K. M. Rabe and D. Vanderbilt
Comp. Mater. Sci., 2014, (81), 446
https://www.sciencedirect.com/science/article/pii/S0927025613005077
18. Orthorhombic ABC semiconductors as antiferroelectrics:
J. W. Bennett, K. F. Garrity, K. M. Rabe, D. Vanderbilt
Phys. Rev. Lett., 2013, (110), 017603
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.110.017603

17. Discovery and design of functional materials: Integration of database searching and first-principles calculations:
J. W. Bennett
Physics Procedia, 2012, (34) 14-23
https://www.sciencedirect.com/science/article/pii/S1875389212013168
16. Integration of first-principles methods and crystallographic database searches for new ferroelectrics: Strategies and explorations:
J. W. Bennett and K.M. Rabe,
J. Solid State Chem., 2012, (195) 21-31
https://www.sciencedirect.com/science/article/pii/S002245961200326X
15. Hexagonal ABC semiconductors as ferroelectrics:
J. W. Bennett, K. F. Garrity, K. M. Rabe and D. Vanderbilt
Phys. Rev. Lett., 2012, (109) 167602-1-4
http://physics.aps.org/synopsis-for/10.1103/PhysRevLett.109.167602

14. Half-Heusler semiconductors as piezoelectrics:
A. Roy, J. W. Bennett, K. M. Rabe and D. Vanderbilt
Phys. Rev. Lett. 2012, (109) 037602
https://journals.aps.org/prl/abstract/10.1103/PhysRevLett.109.037602

Graduate Research Published at the University of Pennsylvania
13. The structural diversity of ABS3 compounds with d0 electronic configuration for the B-cation
J. Brehm, J. W. Bennett, M. R. Schoenberg, I. Grinberg, and A. M. Rappe
J. Chem. Phys., 2014 (140) 224703-1-8
https://aip.scitation.org/doi/abs/10.1063/1.4879659
12. Density functional theory study of PbTiO3-based oxysulfides
J. A. Brehm, H. Takenaka, C.-W. Lee, I. Grinberg, J.W. Bennett, M. R. Schoenberg, and A. M. Rappe
Phys. Rev. B., 2014 (89) 195202-1-8
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.89.195202
11. A first-principles study of band gap engineering via oxygen vacancy doping in ABB’O3 perovskite solid solutions:
T. Qi, M. T. Curnan, S. Kim, J. W. Bennett, I. Grinberg and A. M. Rappe
Phys. Rev. B., 2011, (84), 245206
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.84.245206
10. Post density functional theory studies of highly polar semiconductor PbTi1-xNixO3-z solutions:
G. Y. Gou, J. W. Bennett, H. Takenaka and A. M. Rappe
Phys. Rev. B., 2011, (83) 205115-1-7
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.83.205115
9. Pb-free ferroelectrics investigated with density-functional theory: Sn(Al1/2Nb1/2)O3 perovskites
J. W. Bennett, I. Grinberg, P. K. Davies and A. M. Rappe
Phys. Rev. B., 2011, (83) 144122-1-6
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.83.144112
8. Pb-free semiconductor ferroelectrics: A theoretical study of Pd-substituted Ba(Ti1-xCex)O3 solid solutions:
J. W. Bennett, I. Grinberg, P. K. Davies and A. M. Rappe
Phys. Rev. B, 2010, (82) 184106-1-5
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.82.184106

7. The effect of substituting S for O: The sulfide perovskite BaZrS3 :
J. W. Bennett, I. Grinberg and A. M. Rappe
Phys. Rev. B., 2009, (79) 235115-1-6
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.79.235115

6. New highly polar semi-conductor ferroelectrics through d8-cation O-vacancy doping of PbTiO3 :
J. W. Bennett, I. Grinberg and A. M. Rappe
J. Amer. Chem. Soc., 2008, (130), 17409-17412
https://pubs.acs.org/doi/abs/10.1021/ja8052249

5. Non-monotonic composition dependence of the dielectric response of Ba1-xCaxZrO3 :
J. W. Bennett, I. Grinberg and A. M. Rappe
Chem. Mater., 2008, (20), 5134-5138
https://pubs.acs.org/doi/abs/10.1021/cm800929e
4. BaCe1-xPdxO3 : Redox controlled ingress and egress of palladium in a perovskite:
J. Li, U. G. Singh, J. W. Bennett, K. Page, J. Weaver, J. P. Zhang, T. Proffen,
A. M. Rappe, S. L. Scott and R. Seshadri
Chem. Mater., 2007, (19), 1418-1426
https://pubs.acs.org/doi/abs/10.1021/cm062500i
3. A Pd-doped perovskite catalyst, BaCe1-xPdxO3-z, for CO oxidation:
U. G. Singh, J. Li, J. W. Bennett, A. M. Rappe, R. Seshadri and S. L. Scott
J. Catalysis, 2007, (249), 349-358
https://www.sciencedirect.com/science/article/pii/S0021951707001698
2. Effect of symmetry-lowering on the dielectric response of BaZrO3 :
J. W. Bennett, I. Grinberg and A.M. Rappe
Phys. Rev. B., 2006, (73), 180102(R)
https://journals.aps.org/prb/abstract/10.1103/PhysRevB.73.180102
Undergraduate Research Published at Drexel University
1. Using ice-cooled condensers in chemistry laboratory:
S. Solomon, B. Brook, S, Rutkowsky and J. Bennett
J. Chem. Ed., 2003, (80), 299-301
https://pubs.acs.org/doi/abs/10.1021/ed080p299